DGE分析是使用RNA-seq來檢測穩態下的mRNA表達水平,這一表達水平是通過mRNA的轉錄,加工和降解速度來決定的。但是,RNA-seq也可以用于研究涉及轉錄,翻譯所涉及的過程與動力學特征,這些研究為基因表達提供了新的思路。
基因表達是一個內在的動態過程,但是在檢測復雜轉錄應答的細微以及快速變化或確定不穩定的非編碼RNAs,例如增強子RNAs方面,常規的DGE分析方法就比較受限。RNA-seq可以用于繪制TSSs以及定量新合成的新生RNA,這就可以用來研究RNA動力學。但是,與DGE分析相比,nascent RNA的分析則比較難,因為它們半衰期短,豐度低。因此,為了研究這些動態的重要性,研究者們就開發了多種方法來分析nascent RNA;這些方法揭示了在啟動子處的差異轉錄程度,表明RNA聚合酶II(Pol II)在啟動子附近的暫停是基因表達的關鍵調節步驟,證明了nascent RNA有直接調節轉錄的作用,并表明其序列和結構影響轉錄的延伸,暫停和停頓,以及發揮染色體修飾結合和增強了子的作用。nascent RNA- seq方法旨在區分新近轉錄的RNA和其它RNAs,這些方法可以分為3類:run-on方法,Pol II免疫沉淀法,代謝標記法(FIG. 4)。
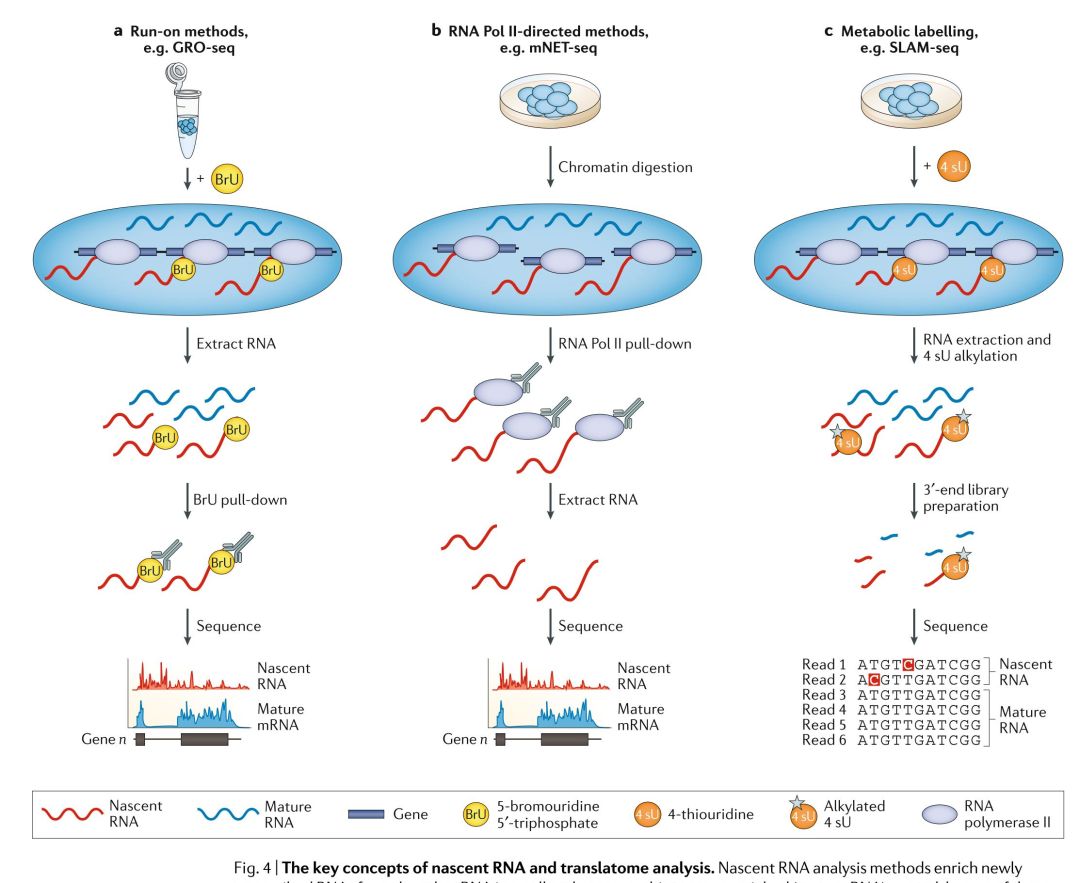
Run-on法是將核酸類似物添加到樣品中,從而使nascent RNA能夠從總的RNA混合物中進行富集,并能夠檢測瞬時RNA的轉錄(FIG. 4a)。全局run-on測序(Global run-on sequencing, GRO-seq)與精確核酸run-on測序(Precision nuclear run-on sequencing, PRO-seq)是分別將Bru或生物素修飾的核酸在RNA的轉錄期整合到nascent RNA中來實現的。其過程大致為,分離細胞核,并通過洗滌除去內源性核苷酸,再添加外源生物素標記的核苷酸,隨后恢復轉錄。通過免疫沉淀或親和純化的方法,對富集的新轉錄RNA進行測序,從而檢測參與轉錄的RNA聚合酶的位置和活性。由于run-on過程中標記的核苷酸的數據,GRO-seq只能測到10-50bp的長度,這就降低的TSS檢測的精度。PRO-seq能夠實現單個堿基級的分辨率,因為生物素標記的核苷酸摻入后轉錄就停止,可以識別出轉錄位點。Run-on方法理解起來很簡單,就是RNA分子整合了修飾的核苷酸,并對其進行富集,用于測序,但是在實踐中,背景中存在有non-nascent RNA,這就需要增加讀長深度。利用這些方法,提示了啟動子處,啟動子處差異或雙向轉錄本起始的程度,確定了增強子RNA在調節基因表達方面的作用。通過特定富集5’加帽的RNAs,GRO-cap,PRO-cap或small 5’capped RNA測序(small 5?-capped RNA sequencing, START-seq)增加了檢測轉錄起始和捕獲RNAs的靈敏度和特異性,這種處理還會降低源于轉錄后加帽的RNAs的背景信號。
Pol II的免疫共沉淀方法包括,天然延長轉錄測序(native elongating transcription sequencing, NET-seq)和哺乳動物染色質天然轉錄測序法(native elongating transcript sequencing for mammalian chromatin, mNET-seq),使用抗FLAG(用FLAG標記的Pol II)抗體進行沉淀的方法,或各種針對Pol II C末端結構域(CTD)的沉淀方法(FIG. 4b)。與這些染色質復合物結合的nascent RNA的RNA-seq方法用于檢測TSSs,雖然non-nascent Pol II結合的RNA與背景mRNA會對讀長濃度產生負面影響,影響分析。NET-seq缺乏特異性,因為任何與Pol II強烈結合的RNA都會污染nascent RNA的富集效果,例如在NET-seq數據中就存在有tRNA和small nucleolar RNA。在mNRET-seq中使用多個CTD抗體提示了VTD修飾是如何影響轉錄的,檢測到了RNA加工的中間體,并能能夠將特定的Pol II nascent RNAs定位于TSSs。然而,這些檢測能力是以更復雜的實驗,更多的細胞數量和更高的測序成本為代價的。
使用核苷酸類似物硫代吡啶(4-thiouridine, 4 sU)進行代謝脈沖標記(Metabolic pulse- labelling)的方法可以識別nascent RNA(FIG. 4c)。但是,在那些需要長標記時間的方法中,大多數的轉錄本都會被標記,這就限制了這種方法的靈敏度。通過專門針對RNAs的3’末端(僅最近拉RNA聚合酶的新轉錄的RNA)的方法,瞬時轉錄組測序(transient transcriptome sequence, TT-seq)與硫醇(SH, thiol)連接的烷基化RNA代謝測序(thiol(SH)-linked alkylation for metabolic sequencing of RNA, SLAM-seq)能夠降低來源于5’RNA的信號。TT-seq將標記時間限制在5分鐘,因此只標記新轉錄本的3’末端,它在進行生物素親和純化前,有一個RNA片段化操作,用于富集標記的RNA。SLAM-seq整合了3’mRNA-seq文庫制備方法(雖然它也用于其它的文庫制備,例如miRNA), 它僅針對標記的新轉錄的RNA進行測序,而非整個轉錄本進行測序。此外,在SLAM-seq中,提取RNA后,還要加入碘乙酰胺(iodoacetamide),用于烷基化已經插入到新生成的nascent RNA鏈中的4 sU殘基。這種修飾會誘導反轉錄式依賴的胞腺嘧啶到胞嘧啶的轉換(T > C),這在測序分析中會被檢測為“突變”,從而直接識別為4 su整合位點。然而,低摻入率意味著只有少量的4 sU位點可以被轉換為胞嘧啶,這就限制了靈敏性。有兩種方法,即TUC-seq與TimeLapse-seq也使用T>C這種突變分析方法,但是它們并不富集3’末端。這兩種方法用于研究細胞干擾后的轉錄應答和RNA的半衰期。
Nascent RNA分析方法還未進行過直接比較。Nascent RNA方法都受到非特異性背景和/或降解的RNA的負面影響,這會影響讀取深度。通過僅測序3’末端,那么non-nascent RNA的效應就會在PRO-seq,TT-seq和SLAM-seq中降低,但是幾乎沒有證據表明是否有其他方法更優。親和純化方法費時費力,與代謝標記法相比,前者需要更多的起始材料,但是,確定脈沖標記的時間比較復雜,并且短脈沖產生用于分析的RNA很少,這限制了靈敏度。最近開發的,組織特異性RNA標記方法以及親折突變分析計算方法或許能夠促進研究者轉向使用生化(基于生物素)富集的手段來研究富含生物學意義的nascent RNA和其它RNA。Nascent RNA方法以及它們與其它方法的隧和,例如空間轉錄組學或RNA-RNA與RNA-蛋白質相互作用的方法,將會提高我們對轉錄過程的理解。