在過去的十年中,RNA測序(RNA-seq)已經成為在全轉錄組范圍內分析差異基因表達和mRNAs差異剪接的重要工具。然而,隨著下一代測序技術的發展,RNA-seq技術也在不斷發展。現在,RNA-seq用于研究RNA生物學的許多方面,其中包括單細胞基因表達、翻譯(翻譯組,translatome)和RNA結構(結構組,structurome)。RNA-seq的其它應用也在開發中,例如空間轉錄學(spatialomics)。加上新的長讀長 (long-read,注:在本文中,RNA-seq測序生成的read統一譯為“讀長“)和直接RNA-seq(direct RNA-seq)技術以及用于數據分析的更好的計算工具的整合,RNA-seq技術的創新有助于人們更全面地理解RNA生物學,例如從何時何地轉錄發生到控制RNA功能的折疊和分子間相互作用等問題。
RNA-seq技術出現于十年之前,自其誕生之日起,RNA-seq就成了研究分子生物學的普遍工具,這項技術幾乎構成了我們對基因組功能的認知基礎 。RNA-seq中最常用的分析方法就是找出差異基因表達(Differential gene expression, DGE)。從最早的出版期刊開始,DGE分析的基本階段就未發生實質性的改變。
在實驗室中,其標準流程就分為三步:
第一步是構建測序文庫,這一步驟包括提取RNA,富集mRNA或清除核糖體RNA,合成 cDNA,加上接頭。
第二步,在高通量平臺(通常是Illumina平臺)上對文庫進行測序,每個樣本的測序深度為10-30M讀長數(讀長這里就是前面說的reads)。
第三步是數據分析,具體的工作是:對測序得到的讀長進行比對(aligning)和/或組裝到轉錄組上,對這些覆蓋了轉錄組的讀長進行過濾,歸一化(Normalization),根據統計模型找出那些在不同樣本之間有差異的轉錄本。早期的RNA-seq從大量的實驗樣本中產生了DGE數據,這充分說明了RNA-seq在廣泛的生物體以及系統中的使用,這些生物體包括玉米(Zea mays), 擬南芥(Arabiodopsis thaliana), 釀酒酵母(Saccharomyces cerevisae),小鼠(Mus musculus)以及人類。雖然RNA-seq這個術語經常被用于那些完全不同的方法學方法和/或生物學,但是DGE分析仍然是RNA-seq(補充材料中的表1)的主要應用,并被視為常規研究工具。
RNA-seq的更廣泛應用已經促進了我們對生物學多方面的理解 ,例如通過提示mRNA剪接和非編碼RNAs和增強子RNAs對基因表達的調控。RNA-seq的應用和進步是由技術發展(濕實驗室和計算生物學)驅動的,相對于以前的基因芯片,RNA-seq這種方法對RNA生物學和轉錄組產生更豐富并且偏見更小的信息。到目前為止,從標準的RNA-seq方法衍生而來的各種RNA-seq方法幾乎有100種。Illumina的短讀長(short-read)測序平臺能對這些由大部分不同方法的RNA-seq構建的文庫進行測序,但是最近長讀長(long-read)RNA-seq的與直接RNA-seq測序(direct RNA sequencing, dRNA-seq)的進步已經能夠解決以前研究人員使用短序列手段無法解決的一些問題。
在這篇綜述中,我們首先會介紹一些最基本的短讀長RNA-seq中的DGE方法,再將這種基礎方法與最近新興的長讀長RNA-seq和dRNA-seq進行比較。我們會介紹短讀長測序方法在文庫制備方面的進展,以及實驗設計和DGE的數據分析方法。隨后我們會拓展這些常規的RNA-seq方法,介紹一些單細胞測序和空間轉錄組學的分析。我們會提供一些案例,介紹RNA-seq在RNA生物學方面的關鍵應用,包括轉錄組分析,翻譯動力學,RNA結構,RNA-RNA之間相互作用和RNA-蛋白質的相互作用。最后,我們會簡單描述一下RNA-seq的未來,以及單細胞和空間RNA-seq方法是否會像DGE分析一樣成為常規工具,長讀長測序方法是否會取代短讀長測序方法。由于篇幅限制,我們無法介紹所有的RNA-seq方法,在這些方法中,值得注意的是非編碼轉錄組學,原核轉錄組學(prokaryotic transcriptomes)和表觀轉錄組學(epitranscriptome)。
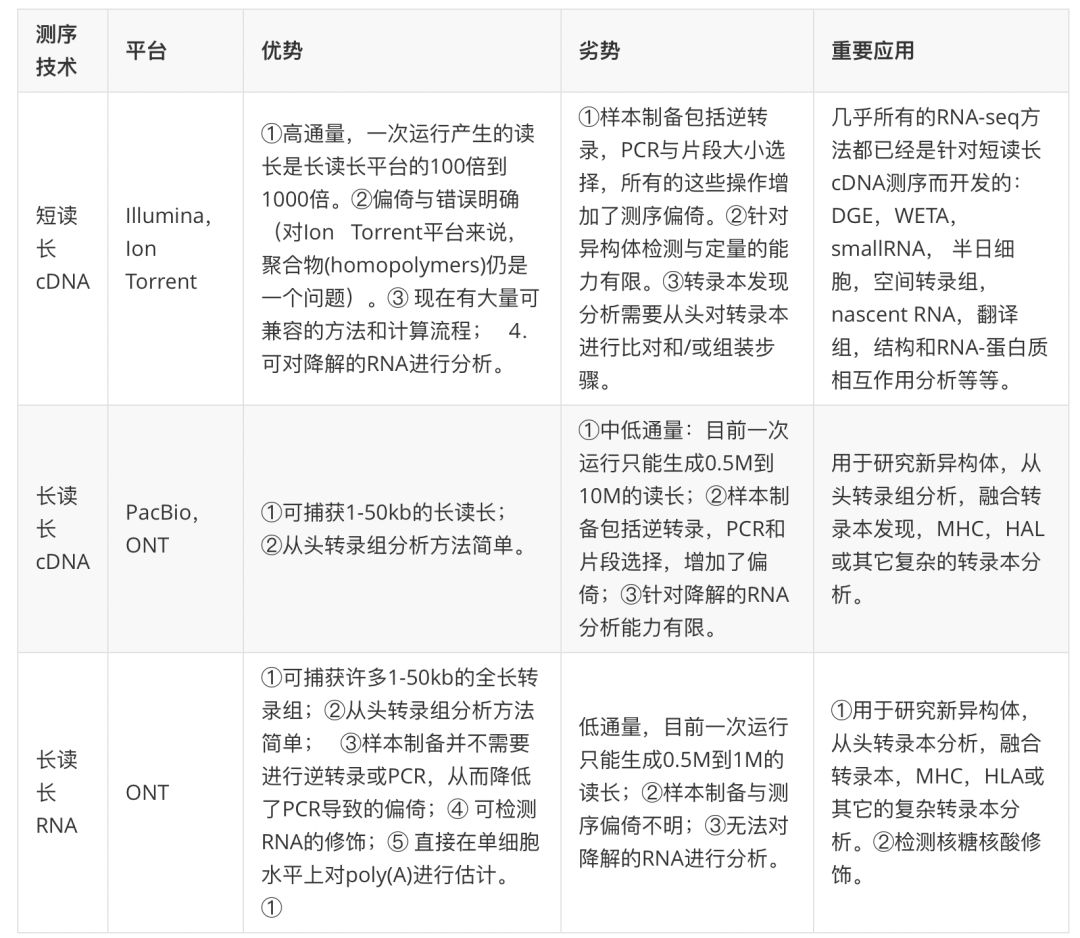
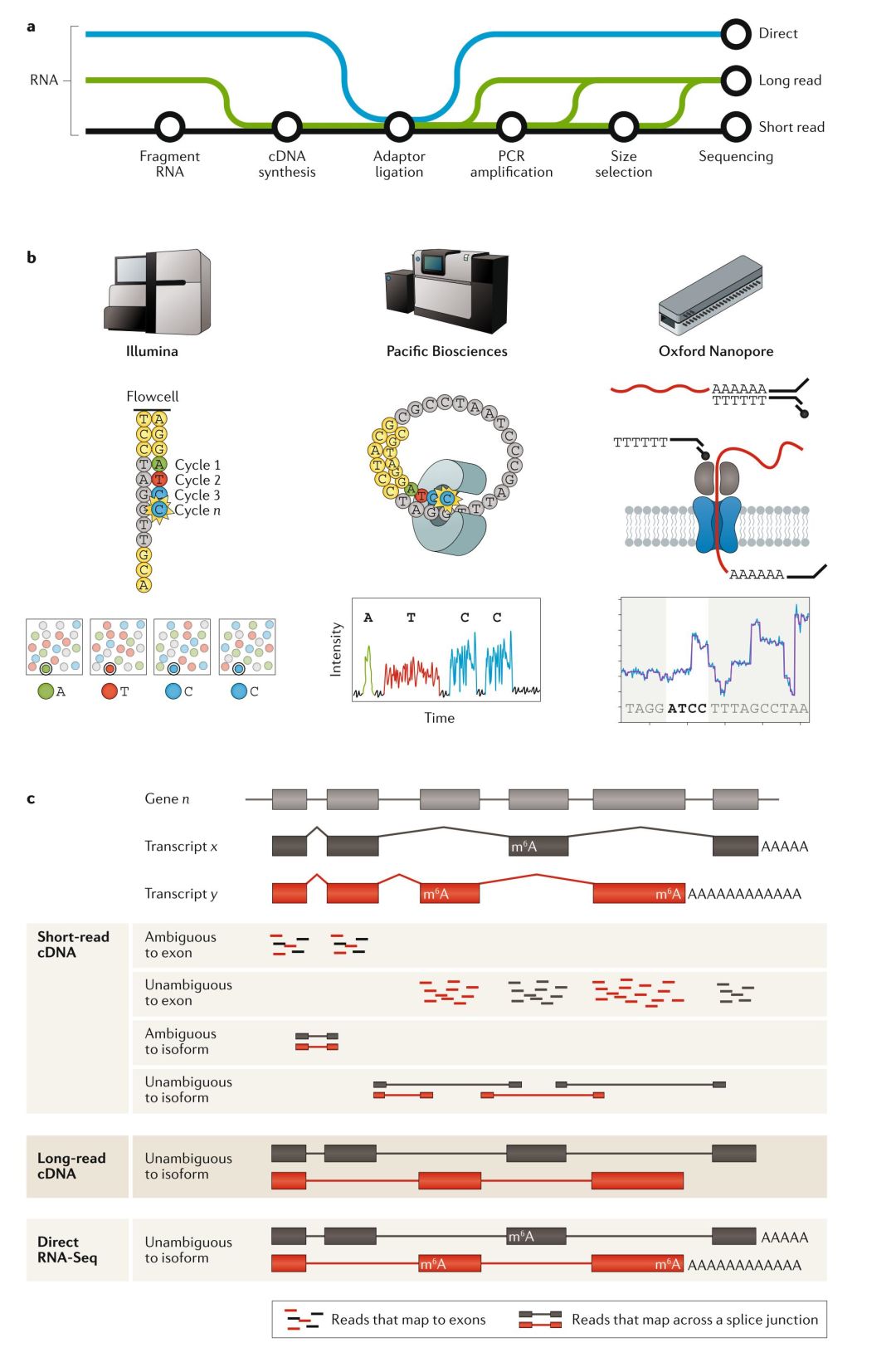
Illumina的短序列讀長測序技術生成了SRA(Short Read Archive)中95%已表達的數據(附件表2)。由于cDNA的短序列讀長測序方法幾乎是一種常規的方法,因此 我們認為這是一種最基礎的 RNA-seq技術,我們先來討論這種測序主要流程與局限。不過,長讀長cDNA測序與dRNA-seq已經興起,隨著研究人員對能提供更豐富轉錄本水平方面(isoform-level)數據需求增大,這兩種新的測序方法有望對常規的短讀長測序方法提出挑戰(FIG1, TABLE1)。
由于高測序錯誤和復雜的基因結構,長讀RNA測序讀段的比對就顯得非常重要。2019年12月16號,哈爾濱工業大學臧天儀、王亞東團隊在GenomeBiology上在線發表了題為deSALT:fastand......
項目負責人YiXing博士是CHOP計算和基因組醫學中心主任,本周他與博士生ZijunZhang和ZhichengPan在《NatureMethods》報道了這款DARTS的框架。DARTS,又稱為深......
柑橘是世界上廣泛種植的果樹作物之一。油斑病是柑橘類水果的一種生理病癥,發生在收獲期和收獲后貯藏期間。目前已在各種柑橘作物中發現這種病癥,對果實質量帶來嚴重影響。油斑病的典型癥狀包括油腺破裂,油腺相鄰的......
全外顯子組測序(WES)和全基因組測序(WGS)對于罕見的孟德爾遺傳病的檢測能力最高只能達到50%,基于這兩種手段,我們對于遺傳多樣性帶來具體生物學功能差異和臨床疾病病因的解釋能力是十分有限的。Cum......