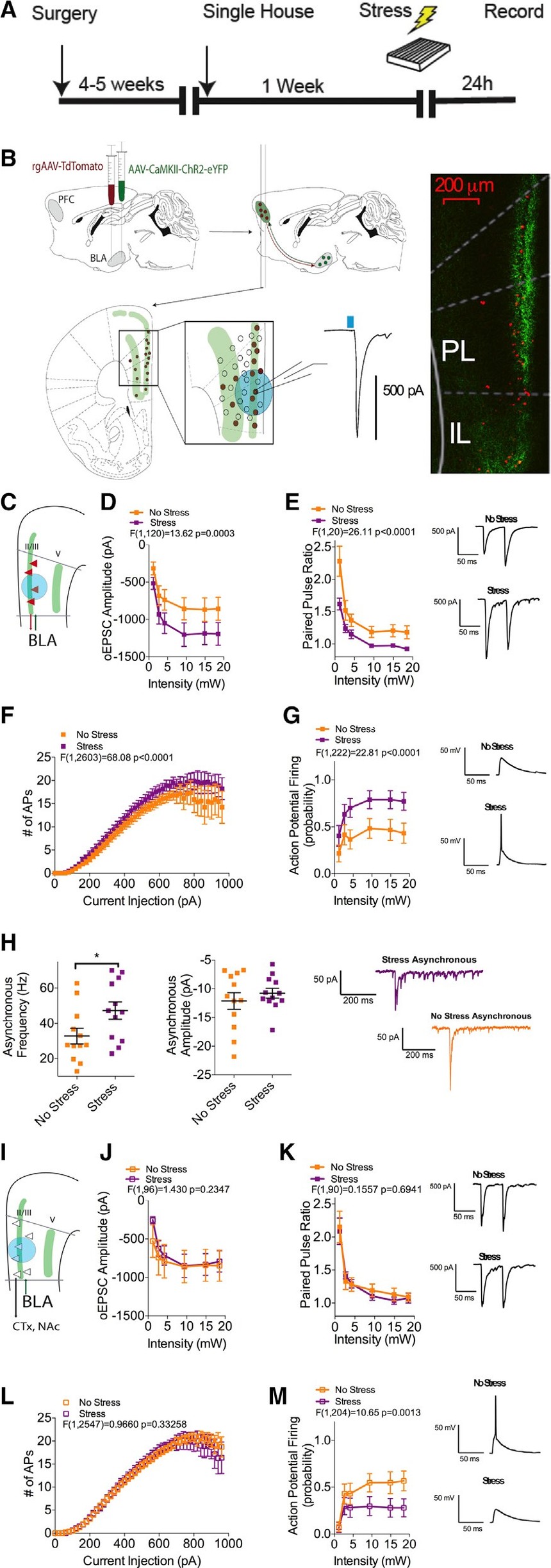
2. 應激暴露增強了BLA-plPFC互反電路中的興奮性信號
數據表明,增強的BLA-plPFC環路活性可能是環境壓力轉化為焦慮樣行為的相關底物。為了研究在BLA-plPFC電路中受到壓力誘導的突觸適應性,使用了順行chr2輔助投射靶向、逆行追蹤方法和體外電生理學相結合的方法(圖2A和2B)。將順行性腺相關病毒aav5 - camki -ChR2- eyfp (ChR2)與逆行性AAV2-CAG-td-Tomato (rAAV)共注射入BLA后4 ~ 6周,處死小鼠進行體外電生理研究。
足底電擊后24小時,研究人員觀察到,rAAV+ L2/3 plPFC神經元中,BLA源性oEPSC振幅增加,配對脈沖比(PPR)下降(圖2C-2E),應激后,在一個互反的BLA-plPFC-BLA子環路中,谷氨酸釋放概率增加。這種增強的興奮性驅動伴隨著rAAV+ L2/3神經元突觸驅動的動作電位放電概率的增加和這些神經元固有的興奮性的少量但顯著的增加(圖2F和2G)。
研究人員還觀察到,在L2/3rAAV+神經元上,光遺傳誘導的異步EPSCs(o-aEPSCs)的頻率增加,但幅度沒有增加,證實了應激暴露后24小時BLA-plPFC突觸的突觸前釋放概率增加(圖2H)。相反,L2/3rAAV神經元的興奮性輸入(圖2J)、PPR(圖2K)或軀體驅動AP放電(圖2L)在應激后沒有明顯變化。在L2/3 rAAV中觀察到的唯一變化是神經元光基因誘發的動作電位放電明顯減少(圖2M)。
3. 內源性大麻素信號廣泛地抑制從BLA到plPFC的谷氨酸能輸入
鑒于觀察到的應激誘導的BLA-plPFC谷氨酸能傳遞的增加是由增強的突觸前釋放介導的,研究人員接下來試圖確定這種效應的驅動機制。眾所周知,逆行作用的ECBs,即anandamide(AEA)和2-AG,受到應激的調節,并調節PFC中的突觸傳遞,這增加了這個神經調制系統中的功能損傷可能有助于應激暴露后BLA-plPFC突觸突觸前驅動增加的可能性。為了驗證這一假設,研究人員首先證明了大麻素受體激動劑cp55,940在rAAV+和rAAV中強烈地抑制了血乳酸誘發的oEPSC振幅。L2/3plPFC神經元,表明BLA投射到plPFC受到大麻素受體的廣泛調節(圖3A、3B、3E和3F)。為了確定ECBs是否調節BLA-plPFC谷氨酸能傳遞,研究人員分析了去極化誘導的興奮抑制(DSE),這是一種典型的2-AG介導的短期突觸抑制,突觸后短暫的去極化導致2-AG的產生,并通過與突觸前CB1受體結合抑制谷氨酸的釋放。研究人員發現,突觸后10s去極化至+30 mV誘導的DSE在L2/3rAAV+和rAAV上均有表達。CB1反向激動劑利莫那班或DO34是2-AG生物合成限速酶(DAGL)的抑制劑(圖3C和3G)。有趣的是,這兩個化合物單獨誘導了PPR的顯著降低,表明2-AG-CB1信號可以緊張性地抑制BLA-plPFC突觸的谷氨酸釋放(圖3D和3H)。為了確保利莫那班對PPR的影響是由于阻斷緊張性ECB信號而不是其反向激動劑特性,研究人員用中性CB1拮抗劑NESS0327(Ness)重復了這一實驗。Ness和利莫那班都阻斷DSE,降低PPR,并導致更大的oEPSC振幅,進一步支持ECB對BLA-plPFC谷氨酸能突觸的緊張性控制。鑒于強直的ECB信號通常被歸因于AEA,而不是2-AG(Kim和Alger,2010),研究人員通過證明利莫那班和DO34都增加了o-aEPSCs的頻率,但沒有增加幅度,從而增強了研究人員的PPR實驗,證實了這些突觸上的強直的2-AG-CB1信號(圖3I-3K)。這些數據為BLAplPFC突觸廣泛表達的多模態強直相2-AG介導的突觸前神經遞質釋放的調節提供了證據。為了檢查ECB信號在plPFC中的輸入特異性,研究人員在檢查MDT向plPFC的投射時進行了相同的關鍵實驗。眾所周知,MDT具有低CB1的表達,因此,研究人員幾乎沒有觀察到CP55,940誘導的oEPSC幅度的抑制和沒有DSE,這表明ECB調節plPFC中的興奮性傳遞具有電路水平的特異性。

4. 應激破壞了對BLA-plPFC互易谷氨酸能回路的2-AG抑制